Genetic ALS / An overview of common mutations
4 min read
SOD1: the “pioneer” genetic mutation
What we now call genetic ALS was first discovered in 1993, when Robert Brown’s research group identified 11 different SOD1 mutations in familial cases of ALS.1,2
Researchers would eventually come to understand that mutant SOD1 proteins can cause excitotoxicity in motor neurons, due to failure in clearing neurotransmitters that trigger motor activity. Over time, hyperexcitable neurons gradually become more vulnerable to motor neuron death, causing the progressive muscle atrophy, spasticity, and fatal respiratory decline seen in ALS.4,5
SOD1 mutations are the second most common mutations among patients with genetic ALS.5 Approximately 187 SOD1 variants have been described in both familial and sporadic cases of ALS to date.
Currently, up to 20% of familial cases (fALS) and 3% of sporadic cases (sALS) have been linked to SOD1 mutations, and these numbers are increasing as systematic genetic screening methods are incorporated into clinical practice.1,6,7


Considerable phenotypic heterogeneity is seen across different SOD1 variants.
For example, the A4V mutation, which is the most frequent variant in North America, gives rise to a fast-progressing form of ALS that typically leads to death within one year from symptom onset. In contrast, the homozygous D90A mutation is associated with an indolent disease course, with patients reaching respiratory failure after 10 or more years of illness.8
SOD1-ALS also manifests in a heterogeneous manner in different parts of the world. The highest frequencies are reported in patients in Japan, Taiwan, and Korea.6
While mutations in the SOD1 gene were the first to be linked to ALS,5 they were hardly the last to help researchers in their pursuit of a deeper understanding of this devastating disease.
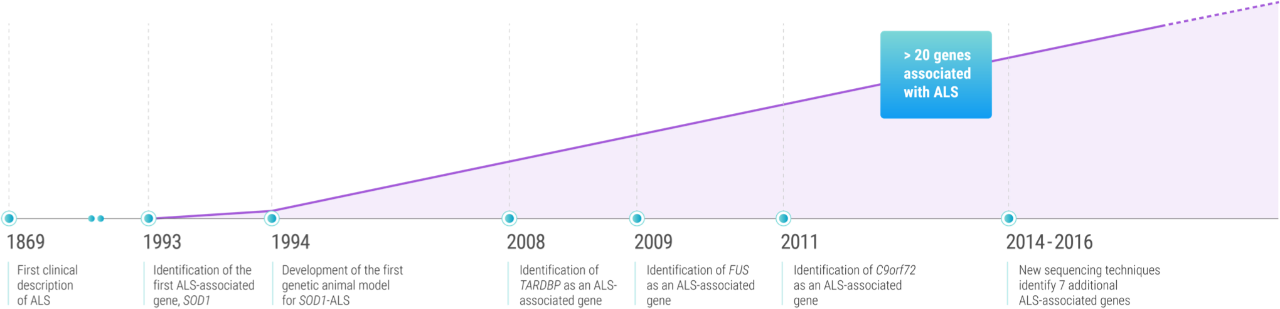
Key milestones in genetic ALS2,5,9-12